Coagulation disorders are a group of rare diseases caused by mutations in blood coagulation factors, which in some cases can manifest with mild symptoms, but in others may involve potentially life-threatening bleeding. The most significant congenital coagulation disorders are: haemophilia A (factor Ⅷ deficiency) and haemophilia B (factor Ⅸ deficiency), with X-linked recessive inheritance, and von Willebrand disease (von Willebrand factor deficiency), with autosomal dominant inheritance in the milder forms, or autosomal recessive inheritance in the more severe forms. All other coagulation factor deficiencies (fibrinogen, prothrombin, factor Ⅴ, factor Ⅶ, factor Ⅹ, factor Ⅺ and factor XⅢ) are much rarer, but even in these cases, they may cause severe or mild bleeding episodes.[1]
The incidence of haemophilia A is 1 in 5,000 males and for haemophilia B it is 1 in 30,000. Patients with mild or moderate haemophilia usually suffer abnormal or excessive bleeding after a traumatic event or surgical intervention, while patients with more severe forms of haemophilia often bleed spontaneously, with subsequent chronic pain and impaired joint mobility. This condition therefore places subjects at risk of potentially fatal bleeding. Therapy for coagulation disorders includes prophylaxis of bleeding events or treatment of an active episode and is based on the intravenous administration of a plasma derivative or a recombinant DNA-product containing the deficient coagulation factor.[2]
More recently, recombinant factors with a prolonged half-life (long-acting) have been synthesized to overcome the difficulties encountered by patients and to improve clinical outcomes; because of their improved pharmacokinetic profile, they make it possible to lengthen the interval between infusions and provide greater safety margins when dealing with bleeding episodes.
In Italy, the National Registry of Congenital Coagulation Disorders collects the data relating to the number and distribution of patients suffering from congenital bleeding disorders in Italy, and also monitors the epidemiology of complications. The current data show that there are 4,109 patients with haemophilia A. Of these, 43.8% suffer the severe form, 13.6% the moderate one and 42.6% the mild form. Eight hundred and eighty-two patients suffer from haemophilia B; of these, 33.9% have a severe form, 21.2% a moderate one and 44.9% a mild form.[3,4]
The graph below shows the epidemiology and prescriptive data for the Region of Umbria (Figure 1 and Table 1), taken from the latest National Report on Congenital Disorders (ISTISAN Reports 20/14).[3]
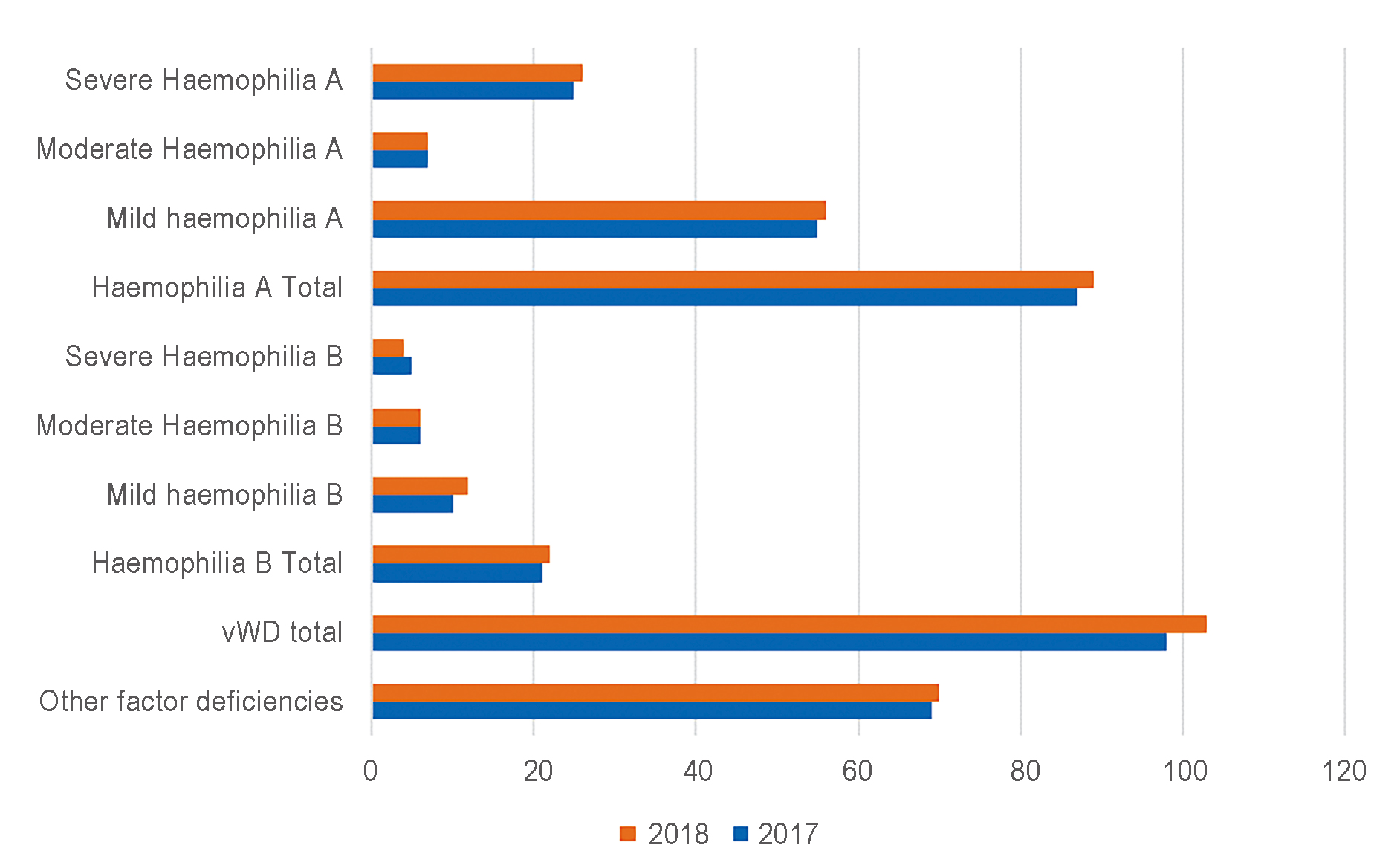
Figure 1: Epidemiological data of haemophilia A and B in Umbria: Number of patients registered.[3] vWD: von Willebrand disease.
| 2017 | 2018 | |
| No. of severe A patients with therapeutic plan | 20 | 24 |
| Percentage of coverage | 80 | 92.3 |
| Fattor Ⅷ prescribed to severe A patients in therapeutic plans (IU) | 5,900,000 | 7,000,000 |
| Total fattor Ⅷ in therapeutic plans (IU) | 7,700,000 | 8,600,000 |
| Plasma derivates (%) | 26.4 | 22.7 |
| Ricombinant (%) | 73.6 | 77.3 |
| 2017 | 2018 | |
| No. of severe B patients with therapeutic plan | 3 | 3 |
| Percentage of coverage | 60 | 75 |
| Fattor Ⅸ prescribed to severe A patients in therapeutic plans (IU) | 900,000 | 700,000 |
| Total fattor Ⅸ in therapeutic plans (IU) | 900,000 | 800,000 |
| Plasma derivates (%) | 19.4 | 19.5 |
| Ricombinant (%) | 80.6 | 80.5 |
In the Region of Umbria, the Regional Reference Centre for haemophiliac patients is the Department of Vascular and Emergency Medicine at the Santa Maria della Misericordia Hospital in Perugia. Approximately 250 haemophiliac patients are monitored at this Operational Unit.
The treatment of haemophilia is strictly home-based, with a therapeutic plan set up by the reference centre and distributed by the patients’ local health unit. However, these patients are at risk of developing internal or external bleeding, both spontaneously and following a trauma, which if not treated promptly could lead to disability or even death.[5]
It is therefore essential for the hospital to have a sufficient supply of the relevant coagulation factors and to guarantee therapy for treating these urgent/emergency episodes, and to manage haemophilia patients who require surgical interventions.
Most of these drugs are very expensive and in the majority of cases are only stored for managing these situations. It is therefore essential to have sufficient supplies to cover the first days/hours of therapy in case of an emergency and to optimise stock in order to avoid waste.
A specific model for hospital management on several levels has been created:
- 1. Individual departments store agreed amounts of these factors. These departments are: the Department of Vascular and Emergency Medicine and the Paediatric Haematology and Oncology Department.
- 2. The Hospital Pharmacy Department stores supplies in the agreed central warehouse in order to guarantee restocking of the department supplies and the management of emergencies.
The National Health System (NHS) pharmacist is an important figure in a patient’s treatment pathway, given that they act as a cornerstone between the requirements of the Operational Units and the sustainability of the actual System. Timely management of stocks makes it possible to handle promptly any emergency situations that may arise, while guaranteeing that the resources available are not wasted.[6] In the current context of major developments in therapeutic innovations in the field of haemophilia, including prolonged half-life concentrates, the roles of the hospital pharmacist and the community pharmaceutical ser vices have become essential for guaranteeing appropriateness, monitoring of therapies and sustainability of innovations with the ultimate goal of generating value for the overall system.[7]
DECLARATIONS
Funding
This research received no external funding.
Conflict of interest
The authors declare no conflict of interest.
REFERENCES
- National Observatory on the Use of Medicines. [The use of Medicines in Italy. National Annual Report 2020.] Rome: Italian Medicines Agency, 2021.
- Srivastava A, Brewer AK, Mauser-Bunschoten EP, Key NS, Kitchen S, Llinas A, et al. Guidelines for the management of hemophilia.
Haemophilia 2013;19:e1-e47. DOI: 10.1111/j.1365-2516.2012.02909.x PMID: 22776238 - Abbonizio F, Arcieri R, Giampaolo A. [Italian Association of Hemophilia Centers (AICE). National Registry of Congenital Coagulation Disorders. 2018 Report (ISTISAN reports 20/14).] Rome: Italian National Institute of Health, 2020.
- Abbonizio F, Giampaolo A, Riccioni R, Arcieri R, Hassan HJ. [Italian Association of Hemophilia Centers (AICE). National Registry ofCongenital Coagulation Disorders. 2016 Report (ISTISAN reports 18/1).] Rome: Italian National Institute of Health, 2018.
- Berntorp E, Andersson NG. Prophylaxis for Hemophilia in the Era of Extended Half-Life Factor VIII/Factor IX Products.
Semin Thromb Hemost 2016;42:518-525. DOI: 10.1055/s-0036-1571315 PMID: 27096762 - Cancanelli L, Mirarchi SA, Pasut E, Salanitro MP, Stoppa S. [The impact of extended half-life factors on FVIII consumption: the hospital pharmacist’s perspective.].
GIHTAD 2018;11:7. - Mannucci PM. Hemophilia therapy: the future has begun.
Haematologica 2020;105:545-553. DOI: 10.3324/haematol.2019.232132 PMID: 32060150